Why natural killer cells?
The most recent (evolutionarily) arm of innate immunity, NK cells, were devised to counter a vulnerability in our immune system. The usual scheme of things - before NK cells - was something like this: you got your B lymphocytes and antibodies to fight off bacteria and ward off parasites. Viruses, however, often found there way inside cells and colonised cells, effectively turning the cell into some primitive version of a zombie, chrning out baby viruses thousands by the minute. Cellular immunity, i.e. T lymphocytes, eventually found a way to deal with those pesky viruses. Viral particles (antigens) were picked up by a molecule called "MHC class I" and presented to T cells, as a distress signal. The T cell could then recognise that particle, and smelling the rot inside, unleashed its destruction machinery on the cell. The cell dies, and so does the virus inside it, everybody loses.
All this was done and dusted very early in evolution though, until some smart teenage mutant ninja virus came up with a devilish idea: why don't attack the cell nucleus and turn off the very expression of MHC class I molecules, effectively rendering the infected cell invisible to T lymphocytes. The virus could then safely reside, and multiply (divide?) in the cell, without being 'seen' by T lymphocytes.
The situation continued for a while, until of course, animals one-upped the virus in the arms race. NK cells appeared first in chordates, as an update patch to plug this very hole. NK cells are large lymphocytes which patrol the body and look for MHC class I molecule. The mechanism is really simple: if a cell does not express MHC class I, NK cells assume the worst (that it's an infected cell, or a cancer cell) and kills the cell with its arsenal of enzymes. Really, a no-brainer solution for smart viruses. This has worked well for us throughout evolution, although indiscriminate killing isn't good strategy in the long term (chronic hepatitis and autoimmune diseases, anyone?), as we're only recently beginning to understand.
 |
| In normal course of things, cytotoxic T cells (CD8+) have no difficulty in identifying virally infected cells because MHC class I molecules bring out those foreign, viral proteins to the surface |
 |
| If a virus has learnt to turn of MHC class I expression, it is invisible to T cells; however, the NK cell notices the absence of MHC class I on the surface, and kills the cell |
Identifying NK cells
NK cells are identified by their low/ variable expression of CD8 and a multifunctional molecule CD56, also called 'neural cell adhesion molecule' (NCAM).
 |
| Initial forward and side scatter plot of peripheral blood mononuclear cells prepared with Lymphoprep; lymphocytes constitute a healthy 29% of mononuclear cells (the debris at the bottom left constitute the other majority) |
 |
| Within the lymphocytes, distribution of CD8 shows three peaks - the negative ones at the left (possibly helper T cells, B cells and macrophages), a dim population in the middle, and a bright one at the right (cytotoxic T cells); the later two constitute 48% of all lymphocytes |
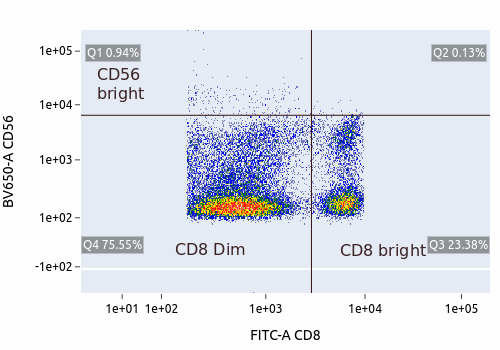 |
| CD56 is expressed at higher levels in the dim population, suggesting that the dim population is where NK cells reside |
 |
| Majority of NK cells express CD56 only dimly (the ones in the box, which turn around to be 20.62% of CD8+ cells, and 9% of all lymphocytes in this particular case). A few of them show bright CD56 (above the box) |
Ref: William G. Morice, MD, PhD. The Immunophenotypic Attributes of NK Cells and NK-Cell Lineage Lymphoproliferative Disorders. (DOI: 10.1309/Q49CRJ030L22MHLF)
Konstantin Khalturin, Matthias Becker, Baruch Rinkevich, Thomas C. G. Bosch. Urochordates and the origin of natural killer cells: Identification of a CD94/NKR-P1-related receptor in blood cells of Botryllus. Proceedings of the National Academy of Sciences Jan 2003, 100 (2) 622-627; DOI: 10.1073/pnas.0234104100




















































