Put simply, as long as we don't find fluorochromes with absolute fidelity (i.e. very narrow emission peaks), we simply have to compensate raw readings from a flow cytometer. Let's start wth just two fluorochromes in two channels,
1. Phycoerythrin (PE) in FL1 detector (red)
2. Fluorescein isothiocynate (FITC) in FL2 detector (yellow)
From a purely theoretical physics perspective, FITC and PE show a consistent and reproducible overlap in their emission spectrum (from Spectra analyser; https://www.biolegend.com/en-us/spectra-analyzer)
 |
| Looking at the two emission spectrum (X axis is wavelength in nm), one can calculate how much FITC emission overlaps with PE, and vice versa; it is evident that FITC has a significant component where PE peak (red) is located, but PE does not have much of a tail where FITC (yellow) peaks |
 |
| By simple eyeballing, one can guess that 15% of the red peak is contributed by the yellow tail, which is very close to the correct figure |
FITCreal = FITCobserved - 0.01 x PEobserved
| FITC | PE | |
| FITC | 1 | 0.15 |
| PE | 0.01 | 1 |
 |
| In this uncompensated graph, find the median fluorescence intensity (MFI) of both FITC and PE channel (although you haven't actually put PE in this tube, just FITC; whatever PE is showing is due to spill from FITC) |
 |
| Note that after compensation, the width of the FITC positive population will increase slightly; this is because the FITC information is now coming from two sources, both FITC and PE, thus increasing its error (degree of freedom); however, the median PE negative line (blue) must remain the same through both populations |

 |
| Capillary electrophoresis of STR PCR; the exact height and area of the peaks are subject to PCR conditions, the loci being chosen and the relative difference in repeats between the two |
 |
| The height/ area of the peaks reflect the relative proportion of the alleles in the mix (remember: all are competing for the same primer!) |
 |
| This is a non-informative STR marker - because the donor and recipient carry the same allele pair |
 |
| The allele at extreme right is ahred between the recipient; the one at extreme left belongs to donor only. Thus, the proportion of the left allele in the final sample is the amount of chimerism |

T lymphocytes don't 'naturally' respond to an antigen; for an antigen to elicit a T cell response
1. A T cell clone with that particular receptor must be present
2. There must be antigen presenting cells (dendritic cells/ macrophages/ B cells)
In vitro, in isolated mononuclear cell preparations from whole blood, the second point is not usually a problem. However, the first condition, presence of antigen specific T cell clone, varies.
For example, for a disease like tuberculosis which is very prevalent in India, one might expect that everyone has at least a few T cell clones for tubercular antigens. However, if the person has never got actual tuberculosis disease, this clone will not be very large. For rare diseases (like Scrub typhus), one would expect an even lower proportion of antigen specific T cells.
Empirically, it is prudent to do a titration of a specific antigen - if only to see which dose does the T cell respond the most. It is not a very tightly controlled experiment, because
1. The total number of lymphocytes vary between people
2. The size of antigen specific T cell clones vary between person to person
3. The T cell response is not dose dependent
However, like all biological phenomenon, T cell stimulation is not an 'all or none' phenomenon, and some correspondence with dose of antigen is always observed. In this case, a whole cell lysate of Mycobacterium tuberculosis (the antigen) was used in increasing doses to stimulate T cells from a healthy person. Production of interferon gamma (IFNG) was used as a marker for T cell response.
1. CD4+ T cells were selected (first panel): the anti-CD4 antibody is tagged with APC
2. The IFNG producing CD4+ T cells are shown in sequence, with increasing doses of the antigen producing a higher proportion of IFNG+ T cells
 |
| The unstimulated population is used as a control to create the box ('gate') for IFNG+ T cells |
The proportion of IFNG+ T cells plateaus at 7.5 microL of antigen dose, and thus this dose was selected.
Thanks Uddeep & Dr Abhinav
Neutrophils (& monocytes) produce hydrogen peroxide (through superoxide production) to kill phagocytosed bacteria.
2O2 + NADPH —> 2O2•– + NADP+ + H+
This reaction is catalaysed by NADPH oxidase. A series of reactions then generates hydrogen peroxide from this superoxide radical, and finally hypochlorous acid.
In vitro, this reaction can be visualised by Dihydrorhodamine (DHR), which is oxidised by H2O2 from normal, stimulated neurophils to give fluorescence (in the FITC channel). EDTA/ heparin blood must be used within 2 hours of collection, in room temparature all the time.
 |
| Gating neutrophils, lymphocytes and monocytes from whole blood |
 |
| Before and after stimulation; note presence of DHR peak ('oxidative burst') in neutrophils and monocytes, and not in lymphocytes |
The lack of superoxide produces a (weirdly named) disease.
Well, this has got (almost) nothing to do with granulomas (maybe a little, indrectly). This is a defect in several components of NADPH oxidase, resulting in inability to produce hydrogen peroxide. An X linked form (75%) & an autosomal recessive form (25%) occur.
XR: Mutation in phagocyte NAPH-O complex due to defect in gene for gp91phox.
AR: Defects in genes p47,67,22 phox or RAC2
It manifests as recurrent pyogenic infection with catalase +ve organisms.
Three scenarios:
1. Normal neutrophils accumulate H2O2 in the phagosome containing ingested bacterium → MPO (myeloperoxidase) is delivered to the phagosome by degranulation, → H2O2 acts as a substrate for MPO to oxidize halide to hypochlorous acid and chloramines, which then kill the microbes. The quantity of hydrogen peroxide produced by normal neutrophils is sufficient to exceed the capacity of catalase, a hydrogen peroxide-catabolizing enzyme of many aerobic microorganisms.
2. When catalase +ve organisms such as E. coli gain entry into the CGD neutrophils, they are not exposed to hydrogen peroxide because the neutrophils do not produce it, and the hydrogen peroxide generated by microbes themselves is destroyed by their own catalase. Thus catalase-positive microbes, such as E. coli, can survive within the phagosome of the CGD neutrophil.
3. When CGD neutrophils ingest catalase negative organisms such streptococci or pneumococci, the organisms generate enough hydrogen peroxide to result in a microbicidal effect; i.e. they are killed by their own hydrogen peroxide.
(Ref: Williams, Hematology)
Thus there is a preponderane by recurrent infections by Catalse positive organisms.
 |
| CGD; note the lack of oxidative burst in stimulated neutrophils |
Titration of FoxP3 (tagged with PE). Note the gradual and slow population shift over increasing doses of antibody. (UU - unstimulated unstained, US - unstimulated stained, SS - stimulated & stained)
 |
| As the dose of antibody is inreased, the entire helper T cell population is taking up the stain (which is unwanted behavior) |
 |
| There is no drop in median PE expression with doses upto 10 microL. This means there might be yet more antibody targets left to bind |
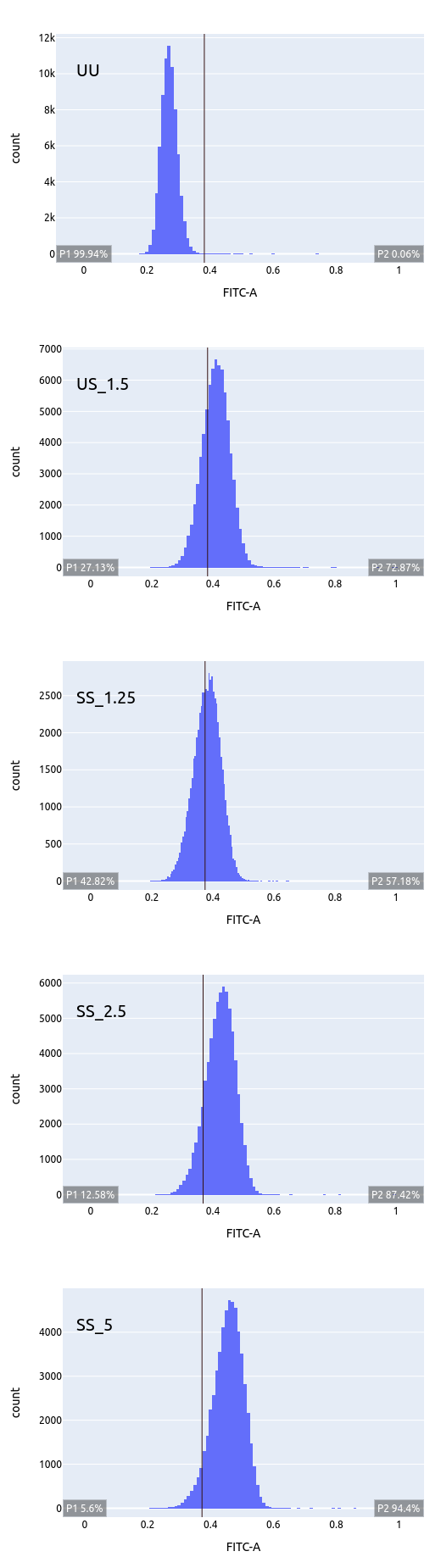
Titration plots for increasing doses of anti-TGFB1. Note the vertical line drawn at unstained (UU) and successive population shifts (non specific binding?). The ideal dose in this case must be close to 0.625 microL.
 |
| Doses ranging from 0.625 to 10 microL; note that entire cell population is moving |
Plotting median FITC versus dose shows a continuously increasing trend. so no help from there!